|
|
|
|
|
|
|
| ||
|
|
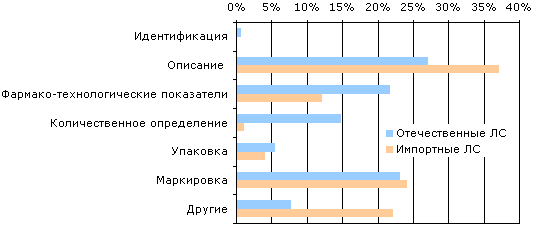
Особенности проведения фармако-технологических испытаний согласно требованиям Государственной фармакопеи УкраиныПод таким названием 24–26 апреля в г. Харькове сотрудниками НЭФЦ и ГНЦЛС был проведен информационно-консультационный семинар для специалистов, занимающихся проведением данных испытаний при разработке, регистрации, производстве, контроле качества лекарственных средств. В докладах, прозвучавших на семинаре, разработчики ГФУ и представители государственных контролирующих органов отразили критические технологические особенности, практические и организационные вопросы проведения, а также перспективы развития обширной группы испытаний лекарственных средств — фармако-технологических тестов, согласно требованиям ГФУ. Фармако-технологические тесты — это тесты, которые так или иначе способны оценить лекарственные препараты как средство доставки некого лекарственного начала к рецептору или мишени в организме,— отметил в докладе Михаил Левин, заведующий отделом ГФУ НЭФЦ. Более подробно он остановился на применении фармако-технологических тестов на различных стадиях фармацевтической разработки, регистрации и рутинном исследовании качества ЛС в развитых странах. Фармако-технологические тесты составляют около 20% от общего числа испытаний в Европейской фармакопее (ЕФ), Британской фармакопее (ВР) и Американской фармакопее (USP). С каждым изданием их абсолютное и относительное число возрастает. В ГФУ вошли 15 общих статей, описывающих проведение фармако-технологических тестов. В докладе ученого секретаря НЭФЦ Аллы Пиотровской и главного специалиста Натальи Крупа, внимание участников семинара акцентировалось на роли фармако-технологических испытаний в фармацевтической разработке с точки зрения ЕС. В основе требований ЕС лежат рекомендации Европейской фармакопеи. Эти требования распространяются на лекарственные препараты, выпускаемые в условиях GМР, и включены в европейскую часть статей ГФУ. Национальная часть ГФУ отражает особенности фармпромышленности Украины, поэтому содержит условия проведения испытаний и рекомендации, отличные от ЕФ, но не противоречащие ей. В ходе фармацевтической разработки отечественные специалисты должны использовать рекомендации, изложенные в национальной части фармако-технологических тестов ГФУ, включая использование оборудования и альтернативных методик этой части. Актуальность проведения семинара была отражена в докладе заместителя Главного государственного инспектора по контролю качества лекарственных средств МЗ Украины Сергея Сура. Он отметил, что фармако-технологические тесты являются важным показателем для оценки как качества ЛС, так и уровня технологии их производства, которые могут быть использованы органами государственного контроля для принятия соответствующих административных решений. В 2001 г. лабораториями территориальных государственных инспекций из проанализированных 76989 образцов отечественных ЛС признаны субстандартными — 1,8%, из проверенных 64144 импортных образцов ЛС — 1,1%. В отличие от импортных ЛС, отечественные препараты часто не соответствовали требованиям АНД именно по фармако-технологическим показателям (рисунок). Это свидетельствует о недостаточно высоком уровне технологии производства и контроля качества ЛС в Украине.
Показатели качества, согласно которым в 2001 г. ЛС не отвечали требованиям АНД (данные лабораторий территориальных госинспекций) Как показывает практика, сама АНД не всегда составлена надлежащим образом. В 2000 г. Центральной лабораторией по контролю качества лекарственных средств выявлены ошибки в 52 ФС и ВФС на отечественные ЛС (около 25% новых АНД), в 2001 г. — в 37 ФС и ВФС на отечественные ЛС и 10 АНД на импортные ЛС. В 2001 г. замечания со стороны Центральной лаборатории по контролю качества лекарственных средств касались таких разделов АНД:
В І квартале 2002 г. при проведении предварительного государственного контроля в Центральной лаборатории по контролю качества лекарственных средств из 74 серий отечественных ЛС 25 (33,8%) от проверенных не отвечали требованиям АНД. При этом 22 серии по фармако-технологическим показателям. Квалификация оборудованияПроблема квалификации оборудования была рассмотрена в докладе руководителя направления «Лекарственные формы и фармако-технологические испытания» отдела ГФУ НЭФЦ Натальи Асмоловой. Квалификация — процесс проверки, подтверждающий, что оборудование работает правильно и его работа приводит к ожидаемым результатам. Квалификация является частью процесса валидации и содержит следующие этапы:
Каждая фирма при поставке определенного прибора предоставляет документацию по инсталляционной, операционной квалификации (IQ, OQ) и квалификации эксплуатационных качеств (PQ). Представителями лаборатории и фирмы-поставщика должны заполняться протоколы по каждому этапу квалификации. Кроме того, для инсталляционного и операционного этапов предоставляется набор инструментов для их проведения. Мероприятия по введению системы квалификации и валидации оборудования в лаборатории могут проводиться в следующих направлениях:
Тест «Растворение» для твердых дозированных лекарственных формНатальей Асмоловой освещены особенности применения теста «Растворение» для определения степени растворения действующих веществ твердых дозированных форм. Тест позволяет получать правильно воспроизводимые результаты с достаточным знанием корреляции данных, полученных из теста «Растворение» и in vivo. Статья ГФУ 2.9.3. «Тест «Растворение» для твердых дозированных форм» гармонизирована со статьей ЕФ и проведение испытания растворения отличается от описанного ГФ ХI. В частности, для проведения теста ГФУ предусматривает использование прибора с лопастью-мешалкой, корзинкой или проточной кюветой. Ранее использовавшийся по ГФ ХI прибор «вращающаяся корзинка» имеет существенные отличия от прибора, описанного ГФУ. Критическими параметрами прибора с корзинкой являются: для сосуда — материал, размеры дна, высота и внутренний диаметр; для корзинки — диаметр проволоки, размеры отверстия, высота сетки; общая высота, внутренний диаметр и внешний диаметр корзинки, внешний диаметр кольца, высота соединяющего диска, позиция перемешивающего устройства, скорость вращения лопасти или корзинки. Одним из остро стоящих вопросов при переходе от требований ГФ ХI к ГФУ по тесту «Растворение» — возможность использование прибора, описанного ГФ ХI, так как для приобретения нового прибора необходимо около 20 тыс. грн. Для конкретных препаратов возможно использовать прибор, описанный в ГФ ХI, после проведения корреляции полученных результатов с прибором ГФУ. Хотя, как показывает практика, использование теста «Растворение» на различных приборах, имеющих по указанным параметрам одинаковые размеры, может приводить к несовпадающим результатам. ГФУ предусматривает отличный от ГФ ХI подход к оценке результатов. Если нет других указаний в частной статье, то для каждой единицы исследуемого препарата за 45 мин. в раствор должно перейти не менее 77% и не более 115% действующих веществ от содержания указанного в разделе «Состав». Если 1 из 6 единиц исследуемого препарата не соответствует требованиям, то проводят исследование растворения еще 6 единиц препарата. Все дополнительные 6 единиц должны соответствовать вышеуказанному требованию. В докладе Дмитрия Леонтьева, руководителя отдела «Валидация и стандартные образцы» отдела ГФУ НЭФЦ описано использование теста «Растворение» при разработке лекарственных средств и на этапе их регистрации. Разработка спецификаций на растворение для оптимизации состава/технологий — совершенно отличная задача от контроля качества ЛС на рынке, так как исследователем целенаправлено создаются максимально чувствительные к изменениям в технологии/составе условия испытания. Например, для хорошо растворимых препаратов используются условия, в которых растворение дает наихудший результат; для плохо растворимых препаратов добиваются приемлемо большего высвобождения. На стадии фармацевтической разработки кроме стандартизированного фармакопейного оборудования возможно использовать любые, в том числе самодельные приборы и устройства для проведения исследований, если они обладают достаточной «дискриминирующей» силой по отношению к препаратам с аномальной биодоступностью. Распадаемость таблеток и капсулПерспективам применения теста «Распадаемость таблеток и капсул» посвящен доклад, подготовленный Натальей Асмоловой и Еленой Выровой, младшим научным сотрудником отдела ГФУ. Наряду с тестом «Растворение» испытание «Распадаемость» является контрольным тестом in vitro, позволяющим судить о доступности действующих веществ лекарственных препаратов, так как в большинстве случаев возможна корреляция скорости растворения с биодоступностью. Под распадаемостью таблеток или капсул подразумевают полное растворение прессованного тела (для растворимых таблеток) или дезагрегацию прессованного тела или капсулы на составляющие частицы гранулята, порошкообразные частицы или мягкую массу, не имеющую ощутимого твердого ядра, в том числе и остатка капсульной оболочки. Время, в течение которого должна распасться соответствующая лекформа, зависит от ее вида. Существенным отличием прибора ГФУ «качающаяся корзинка» от описанного ГФ XI являются различные требования к дискам: согласно ГФУ каждый диск имеет 5 отверстий диаметром 2 мм, согласно ГФ XI — диск без отверстий. Таким образом, результаты испытаний распадаемости, проведенные с применением дисков на приборах по ГФ XI и ГФУ, скорее всего будут различаться. Новым в оценке результатов распадаемости по ГФУ являются следующие аспекты:
Распадаемость суппозиториев и пессариевВ ГФУ введен новый для Украины тест «Распадаемость суппозиториев и пессариев»,— отмечено в докладе Натальи Хованской, зав. Лабораторией фармакопейного анализа, и Натальи Долейко, научного сотрудника Лаборатории фармакопейного анализа. Введение данного теста обусловлено развитием технологий производства суппозиториев и появление суппозиториев на сложных основах, с использованием новых вспомогательных веществ и различных способов введения действующих веществ. Испытанию распадаемости подвергаются все суппозитории, пессарии, капсулы или вагинальные таблетки, кроме предназначенных для модифицированного высвобождения или местного пролонгированного действия, для которых проводиться тест «Растворение». Испытание проводят с помощью прибора, представляющего собой цилиндр, содержащий 2 перфорированных диска на расстоянии 30 мм, помещенный в сосуд с термостатирующим устройством, вместимостью не менее 4 л, заполненный водой. Сосуд снабжен медленно движущейся мешалкой и устройством, поддерживающим прибор в вертикальном положении и позволяющим поворачивать его на 180° не вынимая из воды. Описанный в ГФУ прибор для испытания распадаемости ректальных и вагинальных лекформ предлагается зарубежными поставщиками. По содержащемуся в ГФУ детальному описанию возможно налаживание производства отечественного прибора, что допускается ГФУ. Но при этом основной проблемой является вопрос о квалификации, удостоверяющей, что требования или вспомогательные системы смонтированы должным образом, правильно функционируют и приводят к ожидаемым результатам. Однородность содержания действующего вещества в дозированных лекарственных формахНеобходимость введения в ГФУ теста «Однородность содержания действующего вещества…» обусловлена неравномерностью распределения действующих веществ, что характерно для дозированных ЛС с низким содержанием активного компонента,— подчеркнул в выступлении Александр Гризодуб, заместитель директора НЭФЦ. Причина этого в большинстве случаев связана с технологической невозможностью получить полностью однородный образец. Тест на однородность содержания проводится для всех дозированных лекформ (прессованные таблетки, суппозитории, гранулы, суспензии, лиофилизированные и стерильные рассыпки), исключая дозированные поливитамины и микроэлементы. Тест реализуется методом прямого определения и рассчетно-весовым методом. Статья ГФУ «Однородность содержания действующего вещества…» содержит европейскую часть и национальную. Такой подход обусловлен тем, что ЕФ предусматривает при производстве ЛС соблюдение правил GMP и высокий уровень технологии, обеспечивающий при достаточно низких дозировках действующих веществ однородность дозы. Проверка однородности содержания действующих веществ в единице ЛС становится необходимой по ЕФ (европейская часть ГФУ) при содержании действующих веществ менее 2 мг на единицу ЛС или менее 2% от массы единицы ЛС. Этап 1: пределы отклонений ±15% от средней массы испытуемого образца. Этап 2: пределы отклонений ±25% от средней массы испытуемого образца. В национальной части статьи ГФУ реализован американский подход к определению однородности содержания действующих веществ, так как он наиболее близок к действующей в Украине системе контроля качества и значительно лучше обеспечивают качество ЛС и конкурентоспособность отечественной продукции, при отсутствии GMP ЕС. Пороговая дозировка теста (национальная часть) — 50 мг на единицу ЛС, или 50% от его массы. Этап 1: относительное стандартное отклонение для 10 единиц RSD (10) ≤ 6,0%, пределы отклонений ±15% от значения, указанного в разделе «Состав». Этап 2: относительное стандартное отклонение RSD (30) ≤ 7,8%, пределы отклонений содержания действующего вещества ±25% от значения, указанного в разделе «Состав». Однородность массы для единицы дозированного лекарственного средстваДанная статья является дополнительной к общей статье «Однородность содержания действующего вещества в единице дозированного лекарственного средства» и рассматривается только во взаимосвязи с ней. Тест однородность массы проводится для всех дозированных лекформ, исключая случаи, когда тесту на однородность содержания действующих веществ подвергаются все действующие вещества препарата. Выполняется тест расчетно-весовым методом для 20 единиц препарата. Массы только 2 единиц из 20 могут выходить за пределы отклонения от средней массы, указанной в ГФУ, но ни одна единица не может выходить за интервал, вдвое превышающий эти пределы. В докладах заместителя председателя специализированной экспертной фармацевтической комисии-1 Тамары Согоян «Извлекаемый объем» и научного сотрудника лаборатории инфузионных и ампульных растворов лекарственных средств ГНЦЛС В. Доли «Механические включения (видимые и невидимые частицы, метод микроскопии)» были рассмотрены проблемные вопросы и особенности проведения тестов, применяемых для жидких лекарственных форм для парентерального применения. Доклады старшего научного сотрудника лаборатории аналитической химии НЭФЦ Валентины Чушенко «Ситовой анализ. Определение размера частиц порошков методом микроскопии», зав. лаборатории оптимизации биофармацевтических свойств таблетированных лекарственных средств ГНЦЛС Марка Штейнгардта «Сыпучесть. Насыпной объем. Устойчивость таблеток к раздавливанию» освещали особенности тестов, применяемих при разработке и создании новых ЛС, контроле технологического процесса, входном контроле субстанций и вспомогательных веществ. Круг вопросов, касающихся применения фармако-технологических тестов, использующихся при фармацевтической разработке, производстве, контроле качества мягких лекарственных форм освещен сотрудниками лаборатории жидких и мягких лекформ ГНЦЛС: Николаем Ляпуновым, зав. лабораторией; Еленой Безуглой, с. н. с.; Юрием Столпером, м. н. с. Уделено внимание исследованию реологических свойств мягких лекарственных форм для оптимизации технологии/состава ЛС, в зависимости от желаемого фармакологического действия препарата. В выступлении зав. лабораторией таблеточных лекарственных форм ГНЦЛС Николая Казаринова внимание участников семинара было обращено на различия в национальной и европейской части статьи «Истираемость таблеток без оболочки». Национальная часть ГФУ предусматривает применение прибора, описанного в ГФ XI. В дальнейшем планируется переход на прибор, описанный в европейской части статьи, предусматривающий более жесткие требования к прочности таблеток. Опытом проведения фармако-технологических тестов поделились и. о. зав. Центральной лабораторией по контролю качества лекарственных средств Таисия Герасимчук, зав. Лабораторией фармакопейного анализа НЭФЦ Наталья Хованская, начальник Госииспекции по контролю качества лекарственных средств в Харьковской области Людмила Бондарева. Участники семинара принимали активное участие в обсуждении проблемных вопросов, касающихся проведения фармако-технологических испытаний, делились опытом их проведения на предприятиях и в лабораториях. Непринужденной деловой обстановке и высокой производительности работы специалистов способствовала хорошо продуманная его организаторами программа. Данный семинар является огромным подспорьем специалистам, занимающихся проведением фармако-технологических испытаний при разработке, регистрации, производстве и контроле качества ЛС в повышении качества продукции и уровня технологии производства. Подготовила Мирослава Закотей
© Провизор 1998–2026
|
Грипп. Прививка от гриппа
Нужна ли вакцинация?
Как и чем лечить кашель?
Безрецептурные лекарства при сухом и влажном кашле Устойчивость микробов к антибиотикам →
Помогает ли одежда из шелка лечить экзему?
Что лучше развивает ребёнка — книжки с картинками или с текстом? О безопасности автокресел для детей в возрасте от 4 до 12 лет
Аллергический ринит
Забеременеть в 40 Лечение бесплодия. Обзор существующих вариантов Аденома простаты. Как и чем лечить ? |
|
|
|
|
|